Overview
The skater package provides a collection of analysis and
utility functions for SNP-based
kinship analysis,
testing, and evaluation as an
R package. Functions in the package include tools for
working with pedigree data, performing relationship degree inference,
assessing classification accuracy, and summarizing IBD segment data.
Pedigree parsing and manipulation
Pedigrees define familial relationships in a hierarchical structure.
One of the formats used by PLINK and other genetic analysis tools is
the .fam file.1 A .fam file is a tabular
format with one row per individual and columns for unique IDs of the
mother, father, and the family unit. The package includes
read_fam() to read files in this format:
famfile <- system.file("extdata", "3gens.fam", package="skater", mustWork=TRUE)
fam <- read_fam(famfile)
fam
# # A tibble: 64 × 6
# fid id dadid momid sex affected
# <chr> <chr> <chr> <chr> <int> <int>
# 1 testped1 testped1_g1-b1-s1 0 0 1 1
# 2 testped1 testped1_g1-b1-i1 0 0 2 1
# 3 testped1 testped1_g2-b1-s1 0 0 1 1
# 4 testped1 testped1_g2-b1-i1 testped1_g1-b1-s1 testped1_g1-b1-i1 2 1
# 5 testped1 testped1_g2-b2-s1 0 0 1 1
# 6 testped1 testped1_g2-b2-i1 testped1_g1-b1-s1 testped1_g1-b1-i1 2 1
# 7 testped1 testped1_g3-b1-i1 testped1_g2-b1-s1 testped1_g2-b1-i1 2 1
# 8 testped1 testped1_g3-b2-i1 testped1_g2-b2-s1 testped1_g2-b2-i1 1 1
# 9 testped2 testped2_g1-b1-s1 0 0 2 1
# 10 testped2 testped2_g1-b1-i1 0 0 1 1
# # ℹ 54 more rowsFamily structures imported from “.fam” formated files can then be
translated to the pedigree structure used by the
kinship2 package.2 The “fam” format may include multiple
families, and the fam2ped() function will collapse them all
into a tibble with one row per family:
peds <- fam2ped(fam)
peds
# # A tibble: 8 × 3
# fid data ped
# <chr> <list> <list>
# 1 testped1 <tibble [8 × 5]> <pedigree>
# 2 testped2 <tibble [8 × 5]> <pedigree>
# 3 testped3 <tibble [8 × 5]> <pedigree>
# 4 testped4 <tibble [8 × 5]> <pedigree>
# 5 testped5 <tibble [8 × 5]> <pedigree>
# 6 testped6 <tibble [8 × 5]> <pedigree>
# 7 testped7 <tibble [8 × 5]> <pedigree>
# 8 testped8 <tibble [8 × 5]> <pedigree>In the example above, the resulting tibble is nested by
family ID. The data column contains the individual family
information, while the ped column contains the pedigree
object for that family. You can unnest any particular family:
peds %>%
dplyr::filter(fid=="testped1") %>%
tidyr::unnest(cols=data)
# # A tibble: 8 × 7
# fid id dadid momid sex affected ped
# <chr> <chr> <chr> <chr> <int> <dbl> <list>
# 1 testped1 testped1_g1-b1-s1 NA NA 1 1 <pedigree>
# 2 testped1 testped1_g1-b1-i1 NA NA 2 1 <pedigree>
# 3 testped1 testped1_g2-b1-s1 NA NA 1 1 <pedigree>
# 4 testped1 testped1_g2-b1-i1 testped1_g1-b1-s1 testpe… 2 1 <pedigree>
# 5 testped1 testped1_g2-b2-s1 NA NA 1 1 <pedigree>
# 6 testped1 testped1_g2-b2-i1 testped1_g1-b1-s1 testpe… 2 1 <pedigree>
# 7 testped1 testped1_g3-b1-i1 testped1_g2-b1-s1 testpe… 2 1 <pedigree>
# 8 testped1 testped1_g3-b2-i1 testped1_g2-b2-s1 testpe… 1 1 <pedigree>You can also look at a single pedigree:
peds$ped[[1]]
# Pedigree object with 8 subjects
# Bit size= 4Or plot that pedigree:
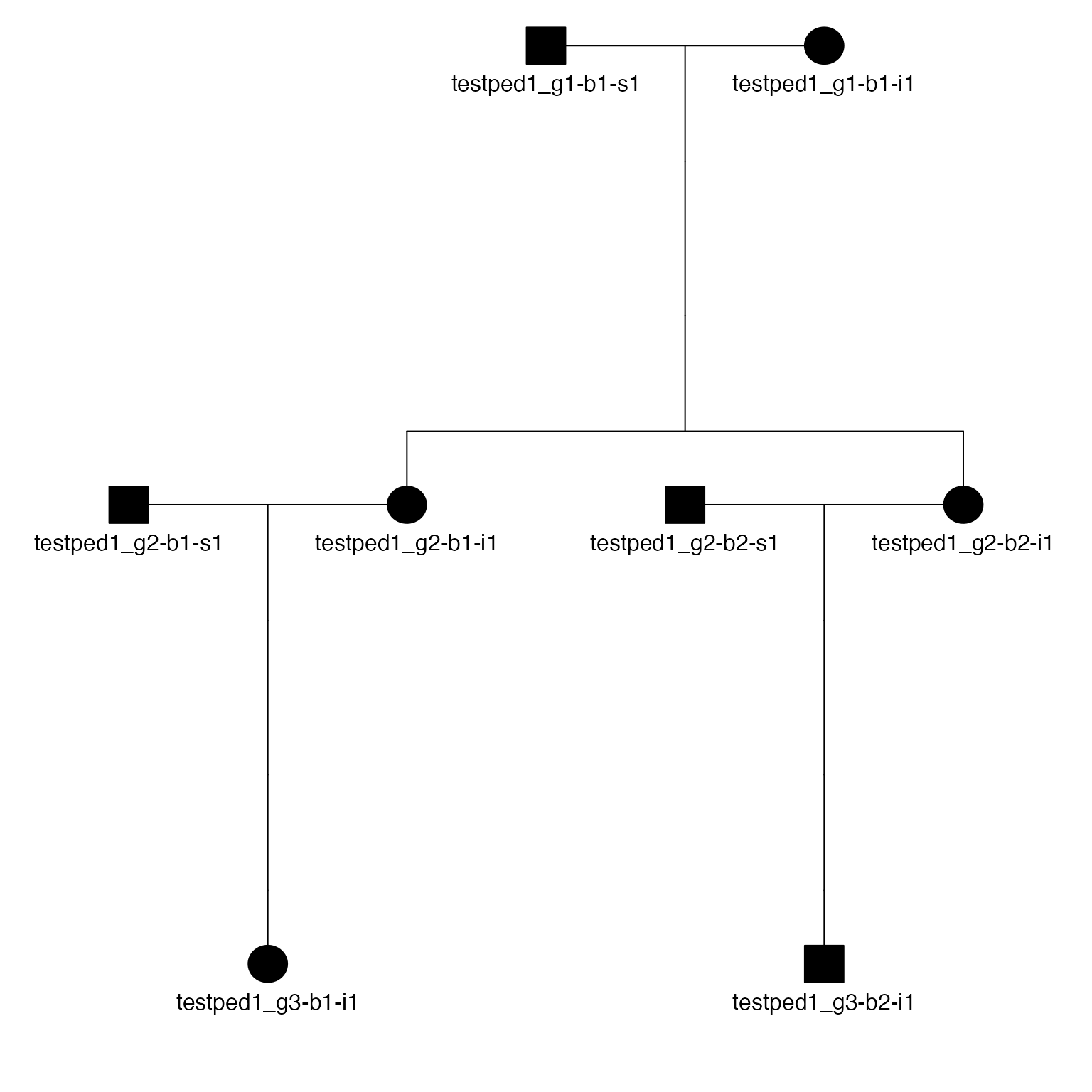
The plot_pedigree() function from skater
will iterate over a list of pedigree objects, writing a multi-page PDF,
with each page containing a pedigree from family:
plot_pedigree(peds$ped, file="3gens.ped.pdf")The ped2kinpair() function takes a pedigree object and
produces a pairwise list of relationships between all individuals in the
data with the expected kinship coefficients for each pair.
The function can be run on a single family:
ped2kinpair(peds$ped[[1]])
# # A tibble: 36 × 3
# id1 id2 k
# <chr> <chr> <dbl>
# 1 testped1_g1-b1-s1 testped1_g1-b1-s1 0.5
# 2 testped1_g1-b1-i1 testped1_g1-b1-s1 0
# 3 testped1_g1-b1-s1 testped1_g2-b1-s1 0
# 4 testped1_g1-b1-s1 testped1_g2-b1-i1 0.25
# 5 testped1_g1-b1-s1 testped1_g2-b2-s1 0
# 6 testped1_g1-b1-s1 testped1_g2-b2-i1 0.25
# 7 testped1_g1-b1-s1 testped1_g3-b1-i1 0.125
# 8 testped1_g1-b1-s1 testped1_g3-b2-i1 0.125
# 9 testped1_g1-b1-i1 testped1_g1-b1-i1 0.5
# 10 testped1_g1-b1-i1 testped1_g2-b1-s1 0
# # ℹ 26 more rowsOr mapped over all families in the pedigree
kinpairs <-
peds %>%
dplyr::mutate(pairs=purrr::map(ped, ped2kinpair)) %>%
dplyr::select(fid, pairs) %>%
tidyr::unnest(cols=pairs)
kinpairs
# # A tibble: 288 × 4
# fid id1 id2 k
# <chr> <chr> <chr> <dbl>
# 1 testped1 testped1_g1-b1-s1 testped1_g1-b1-s1 0.5
# 2 testped1 testped1_g1-b1-i1 testped1_g1-b1-s1 0
# 3 testped1 testped1_g1-b1-s1 testped1_g2-b1-s1 0
# 4 testped1 testped1_g1-b1-s1 testped1_g2-b1-i1 0.25
# 5 testped1 testped1_g1-b1-s1 testped1_g2-b2-s1 0
# 6 testped1 testped1_g1-b1-s1 testped1_g2-b2-i1 0.25
# 7 testped1 testped1_g1-b1-s1 testped1_g3-b1-i1 0.125
# 8 testped1 testped1_g1-b1-s1 testped1_g3-b2-i1 0.125
# 9 testped1 testped1_g1-b1-i1 testped1_g1-b1-i1 0.5
# 10 testped1 testped1_g1-b1-i1 testped1_g2-b1-s1 0
# # ℹ 278 more rowsNote that this maps ped2kinpair() over all
ped objects in the input tibble, and that
relationships are not shown for between-family relationships (which
should all be zero).
Degree Inference
The skater package includes functions to translate
kinship coefficients to relationship degrees. The kinship coefficients
could come from ped2kinpair() or other kinship estimation
software.
The dibble() function creates a degree
inference tibble, with degrees up to the
specified max_degree (default=3), expected kinship
coefficient, and lower (l) and upper (u)
inference ranges as defined in the KING paper.3 Degree 0 corresponds
to self / identity / monozygotic twins, with an expected kinship
coefficient of 0.5, with inference range >=0.354. Anything beyond the
maximum degree resolution is considered unrelated (degree
NA), with expected kinship coefficient of 0.
dibble()
# # A tibble: 5 × 4
# degree k l u
# <int> <dbl> <dbl> <dbl>
# 1 0 0.5 0.354 1
# 2 1 0.25 0.177 0.354
# 3 2 0.125 0.0884 0.177
# 4 3 0.0625 0.0442 0.0884
# 5 NA 0 -1 0.0442The degree inference max_degree default is 3. Change
this argument to allow more granular degree inference ranges:
dibble(max_degree = 5)
# # A tibble: 7 × 4
# degree k l u
# <int> <dbl> <dbl> <dbl>
# 1 0 0.5 0.354 1
# 2 1 0.25 0.177 0.354
# 3 2 0.125 0.0884 0.177
# 4 3 0.0625 0.0442 0.0884
# 5 4 0.0312 0.0221 0.0442
# 6 5 0.0156 0.0110 0.0221
# 7 NA 0 -1 0.0110Note that the distance between relationship degrees becomes smaller
as the relationship degree becomes more distant. The
dibble() function will throw a warning with
max_degree >=10, and will stop with an error at
>=12.
The kin2degree() function infers the relationship degree
given a kinship coefficient and a max_degree up to which
anything more distant is treated as unrelated. Example first degree
relative:
kin2degree(.25, max_degree=3)
# [1] 1Example 4th degree relative, but using the default max_degree resolution of 3:
kin2degree(.0312, max_degree=3)
# [1] NAExample 4th degree relative, but increasing the degree resolution:
kin2degree(.0312, max_degree=5)
# [1] 4The kin2degree() function is vectorized over values of
k, so it can be used inside of a mutate on a
tibble of kinship coefficients:
# Get two pairs from each type of relationship we have in kinpairs:
kinpairs_subset <-
kinpairs %>%
dplyr::group_by(k) %>%
dplyr::slice(1:2)
kinpairs_subset
# # A tibble: 10 × 4
# # Groups: k [5]
# fid id1 id2 k
# <chr> <chr> <chr> <dbl>
# 1 testped1 testped1_g1-b1-i1 testped1_g1-b1-s1 0
# 2 testped1 testped1_g1-b1-s1 testped1_g2-b1-s1 0
# 3 testped1 testped1_g3-b1-i1 testped1_g3-b2-i1 0.0625
# 4 testped2 testped2_g3-b1-i1 testped2_g3-b2-i1 0.0625
# 5 testped1 testped1_g1-b1-s1 testped1_g3-b1-i1 0.125
# 6 testped1 testped1_g1-b1-s1 testped1_g3-b2-i1 0.125
# 7 testped1 testped1_g1-b1-s1 testped1_g2-b1-i1 0.25
# 8 testped1 testped1_g1-b1-s1 testped1_g2-b2-i1 0.25
# 9 testped1 testped1_g1-b1-s1 testped1_g1-b1-s1 0.5
# 10 testped1 testped1_g1-b1-i1 testped1_g1-b1-i1 0.5
# Infer degree out to third degree relatives:
kinpairs_subset %>%
dplyr::mutate(degree=kin2degree(k, max_degree=3))
# # A tibble: 10 × 5
# # Groups: k [5]
# fid id1 id2 k degree
# <chr> <chr> <chr> <dbl> <int>
# 1 testped1 testped1_g1-b1-i1 testped1_g1-b1-s1 0 NA
# 2 testped1 testped1_g1-b1-s1 testped1_g2-b1-s1 0 NA
# 3 testped1 testped1_g3-b1-i1 testped1_g3-b2-i1 0.0625 3
# 4 testped2 testped2_g3-b1-i1 testped2_g3-b2-i1 0.0625 3
# 5 testped1 testped1_g1-b1-s1 testped1_g3-b1-i1 0.125 2
# 6 testped1 testped1_g1-b1-s1 testped1_g3-b2-i1 0.125 2
# 7 testped1 testped1_g1-b1-s1 testped1_g2-b1-i1 0.25 1
# 8 testped1 testped1_g1-b1-s1 testped1_g2-b2-i1 0.25 1
# 9 testped1 testped1_g1-b1-s1 testped1_g1-b1-s1 0.5 0
# 10 testped1 testped1_g1-b1-i1 testped1_g1-b1-i1 0.5 0Benchmarking Degree Classification
Once estimated kinship is converted to degree, it may be of interest to compare the inferred degree to truth. When aggregated over many relationships and inferences, this method can help benchmark performance of a particular kinship analysis method.
The skater package adapts functionality from the
confusionMatrix package4 in the confusion_matrix()
function.
The confusion_matrix() function on its own outputs a
list with three objects:
- A
tibblewith calculated accuracy, lower and upper bounds, the guessing rate and p-value of the accuracy vs. the guessing rate. - A
tibblewith the following statistics (for each class):- Sensitivity = A/(A+C)
- Specificity = D/(B+D)
- Prevalence = (A+C)/(A+B+C+D)
- PPV = (sensitivity * prevalence)/((sensitivity * prevalence) + ((1-specificity) * (1-prevalence)))
- NPV = (specificity * (1-prevalence))/(((1-sensitivity) * prevalence) + ((specificity) * (1-prevalence)))
- Detection Rate = A/(A+B+C+D)
- Detection Prevalence = (A+B)/(A+B+C+D)
- Balanced Accuracy = (sensitivity+specificity)/2
- Precision = A/(A+B)
- Recall = A/(A+C)
- F1 = harmonic mean of precision and recall
- False Discovery Rate = 1 - PPV
- False Omission Rate = 1 - NPV
- False Positive Rate = 1 - Specificity
- False Negative Rate = 1 - Sensitivity
- A
matrixwith the contingency table object itself. - A
vectorwith the reciprocal RMSE (R-RMSE). The R-RMSE is calculated assqrt(mean((1/(Target+.5)-1/(Predicted+.5))^2))), and is a superior measure to classification accuracy when benchmarking relationship degree estimation. Taking the reciprocal of the target and predicted degree results in larger penalties for more egregious misclassifications (e.g., classifying a first-degree relative pair as second degree) than misclassifications at more distant relationships (e.g., misclassifying a fourth-degree relative pair as fifth-degree). The +0.5 adjustment prevents division-by-zero when a 0th-degree (identical) relative pair is introduced.
To illustrate the usage, first take the kinpairs data
from above and randomly flip ~20% of the true relationship degrees.
# Function to randomly flip levels of a factor (at 20%, by default)
randomflip <- function(x, p=.2) ifelse(runif(length(x))<p, sample(unique(x)), x)
# Infer degree (truth/target) using kin2degree, then randomly flip 20% of them
set.seed(42)
kinpairs_inferred <- kinpairs %>%
dplyr::mutate(degree_truth=kin2degree(k, max_degree=3)) %>%
dplyr::mutate(degree_truth=as.character(degree_truth)) %>%
dplyr::mutate(degree_truth=tidyr::replace_na(degree_truth, "unrelated")) %>%
dplyr::mutate(degree_inferred=randomflip(degree_truth))
kinpairs_inferred
# # A tibble: 288 × 6
# fid id1 id2 k degree_truth degree_inferred
# <chr> <chr> <chr> <dbl> <chr> <chr>
# 1 testped1 testped1_g1-b1-s1 testped1_g1-b1… 0.5 0 0
# 2 testped1 testped1_g1-b1-i1 testped1_g1-b1… 0 unrelated unrelated
# 3 testped1 testped1_g1-b1-s1 testped1_g2-b1… 0 unrelated unrelated
# 4 testped1 testped1_g1-b1-s1 testped1_g2-b1… 0.25 1 1
# 5 testped1 testped1_g1-b1-s1 testped1_g2-b2… 0 unrelated unrelated
# 6 testped1 testped1_g1-b1-s1 testped1_g2-b2… 0.25 1 1
# 7 testped1 testped1_g1-b1-s1 testped1_g3-b1… 0.125 2 2
# 8 testped1 testped1_g1-b1-s1 testped1_g3-b2… 0.125 2 1
# 9 testped1 testped1_g1-b1-i1 testped1_g1-b1… 0.5 0 0
# 10 testped1 testped1_g1-b1-i1 testped1_g2-b1… 0 unrelated unrelated
# # ℹ 278 more rows
confusion_matrix(prediction = kinpairs_inferred$degree_inferred,
target = kinpairs_inferred$degree_truth)
# $Accuracy
# # A tibble: 1 × 5
# Accuracy `Accuracy LL` `Accuracy UL` `Accuracy Guessing` `Accuracy P-value`
# <dbl> <dbl> <dbl> <dbl> <dbl>
# 1 0.812 0.763 0.856 0.333 1.09e-62
#
# $Other
# # A tibble: 6 × 15
# Class N Sensitivity/Recall/T…¹ `Specificity/TNR` `PPV/Precision` NPV
# <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
# 1 0 64 0.75 0.964 0.857 0.931
# 2 1 72 0.806 0.944 0.829 0.936
# 3 2 48 0.833 0.967 0.833 0.967
# 4 3 8 0.75 0.936 0.25 0.992
# 5 unrelated 96 0.854 0.958 0.911 0.929
# 6 Average 57.6 0.799 0.954 0.736 0.951
# # ℹ abbreviated name: ¹`Sensitivity/Recall/TPR`
# # ℹ 9 more variables: `F1/Dice` <dbl>, Prevalence <dbl>,
# # `Detection Rate` <dbl>, `Detection Prevalence` <dbl>,
# # `Balanced Accuracy` <dbl>, FDR <dbl>, FOR <dbl>, `FPR/Fallout` <dbl>,
# # FNR <dbl>
#
# $Table
# Target
# Predicted 0 1 2 3 unrelated
# 0 48 4 2 1 1
# 1 5 58 4 0 3
# 2 0 3 40 1 4
# 3 8 4 0 6 6
# unrelated 3 3 2 0 82
#
# $recip_rmse
# [1] 0.4665971You can use purrr::pluck() to isolate just the
contingency table:
confusion_matrix(prediction = kinpairs_inferred$degree_inferred,
target = kinpairs_inferred$degree_truth) %>%
purrr::pluck("Table")
# Target
# Predicted 0 1 2 3 unrelated
# 0 48 4 2 1 1
# 1 5 58 4 0 3
# 2 0 3 40 1 4
# 3 8 4 0 6 6
# unrelated 3 3 2 0 82Or optionally output in a tidy (longer=TRUE) format,
then spread stats by class:
confusion_matrix(prediction = kinpairs_inferred$degree_inferred,
target = kinpairs_inferred$degree_truth,
longer = TRUE) %>%
purrr::pluck("Other") %>%
tidyr::spread(Class, Value) %>%
dplyr::relocate(Average, .after=dplyr::last_col()) %>%
dplyr::mutate_if(rlang::is_double, signif, 2) %>%
knitr::kable()| Statistic | 0 | 1 | 2 | 3 | unrelated | Average |
|---|---|---|---|---|---|---|
| Balanced Accuracy | 0.860 | 0.880 | 0.900 | 0.8400 | 0.910 | 0.880 |
| Detection Prevalence | 0.190 | 0.240 | 0.170 | 0.0830 | 0.310 | 0.200 |
| Detection Rate | 0.170 | 0.200 | 0.140 | 0.0210 | 0.280 | 0.160 |
| F1/Dice | 0.800 | 0.820 | 0.830 | 0.3800 | 0.880 | 0.740 |
| FDR | 0.140 | 0.170 | 0.170 | 0.7500 | 0.089 | 0.260 |
| FNR | 0.250 | 0.190 | 0.170 | 0.2500 | 0.150 | 0.200 |
| FOR | 0.069 | 0.064 | 0.033 | 0.0076 | 0.071 | 0.049 |
| FPR/Fallout | 0.036 | 0.056 | 0.033 | 0.0640 | 0.042 | 0.046 |
| N | 64.000 | 72.000 | 48.000 | 8.0000 | 96.000 | 58.000 |
| NPV | 0.930 | 0.940 | 0.970 | 0.9900 | 0.930 | 0.950 |
| PPV/Precision | 0.860 | 0.830 | 0.830 | 0.2500 | 0.910 | 0.740 |
| Prevalence | 0.220 | 0.250 | 0.170 | 0.0280 | 0.330 | 0.200 |
| Sensitivity/Recall/TPR | 0.750 | 0.810 | 0.830 | 0.7500 | 0.850 | 0.800 |
| Specificity/TNR | 0.960 | 0.940 | 0.970 | 0.9400 | 0.960 | 0.950 |
IBD Segment Analysis
Tools such as hap-ibd5 are capable of inferring shared IBD
segments between individuals. The skater package includes
functionality to take those IBD segments, compute shared genomic
centimorgan (cM) length, and convert that shared cM to a kinship
coefficient. In addition to inferred segments, these functions can
estimate “truth” kinship from data simulated by ped-sim.6
The read_ibd() function reads in the pairwise IBD
segment format. Input to this function can either be inferred IBD
segments from hap-IBD (source="hapibd") or simulated
segments (source="pedsim"). The first example below uses
data in the hap-ibd output format:
hapibd_fp <- system.file("extdata", "GBR.sim.ibd.gz", package="skater", mustWork=TRUE)
hapibd_seg <- read_ibd(hapibd_fp, source = "hapibd")
# New names:
# • `1` -> `1...2`
# • `1` -> `1...4`
# • `1` -> `1...5`
hapibd_seg
# # A tibble: 3,954 × 6
# id1 id2 chr start end length
# <chr> <chr> <dbl> <dbl> <dbl> <dbl>
# 1 testped1_g1-b1-s1 testped1_g3-b1-i1 1 197661576 234863602 47.1
# 2 testped1_g2-b2-i1 testped1_g3-b1-i1 1 197661576 231017545 39.8
# 3 testped1_g3-b1-i1 testped1_g3-b2-i1 1 197661576 212799139 20.3
# 4 testped3_g1-b1-s1 testped3_g3-b2-i1 1 2352146 10862397 17.7
# 5 testped3_g2-b2-i1 testped3_g3-b2-i1 1 2352146 10862397 17.7
# 6 testped1_g1-b1-s1 testped1_g2-b1-i1 1 3328659 64123868 86.4
# 7 testped1_g1-b1-s1 testped1_g3-b1-i1 1 3328659 33476811 51.2
# 8 testped1_g2-b2-s1 testped1_g3-b2-i1 1 5003504 32315147 45.9
# 9 testped2_g1-b1-i1 testped2_g3-b1-i1 1 240810528 248578622 15.9
# 10 testped2_g1-b1-i1 testped2_g2-b2-i1 1 241186056 249170711 15.5
# # ℹ 3,944 more rowsIn order to translate the shared genomic cM length to a kinship
coefficient, you must load a genetic map with read_map().
Software for IBD segment inference and simulation requires a genetic
map. The map loaded for kinship estimation should be the same one used
for creating the shared IBD segment output. The example below uses a
minimal genetic map created with min_map7 that ships with
skater:
gmapfile <- system.file("extdata", "sexspec-avg-min.plink.map", package="skater", mustWork=TRUE)
gmap <- read_map(gmapfile)
gmap
# # A tibble: 28,726 × 3
# chr value bp
# <dbl> <dbl> <dbl>
# 1 1 0 752721
# 2 1 0.0292 1066029
# 3 1 0.0829 1099342
# 4 1 0.157 1106473
# 5 1 0.246 1152631
# 6 1 0.294 1314015
# 7 1 0.469 1510801
# 8 1 0.991 1612540
# 9 1 1.12 1892325
# 10 1 1.41 1916587
# # ℹ 28,716 more rowsThe ibd2kin() function takes the segments and map file
and outputs a tibble with one row per pair of individuals
and columns for individual 1 ID, individual 2 ID, and the kinship
coefficient for the pair:
ibd_dat <- ibd2kin(.ibd_data=hapibd_seg, .map=gmap)
ibd_dat
# # A tibble: 196 × 3
# id1 id2 kinship
# <chr> <chr> <dbl>
# 1 testped1_g1-b1-i1 testped1_g1-b1-s1 0.000316
# 2 testped1_g1-b1-i1 testped1_g2-b1-i1 0.261
# 3 testped1_g1-b1-i1 testped1_g2-b2-i1 0.263
# 4 testped1_g1-b1-i1 testped1_g2-b2-s1 0.000150
# 5 testped1_g1-b1-i1 testped1_g3-b1-i1 0.145
# 6 testped1_g1-b1-i1 testped1_g3-b2-i1 0.133
# 7 testped1_g1-b1-i1 testped2_g1-b1-i1 0.000165
# 8 testped1_g1-b1-i1 testped2_g1-b1-s1 0.000323
# 9 testped1_g1-b1-i1 testped2_g2-b1-i1 0.000499
# 10 testped1_g1-b1-i1 testped2_g2-b1-s1 0.000318
# # ℹ 186 more rowsAs noted above, the IBD segment kinship estimation can be performed on simulated segments. The package includes an example of IBD data in that format:
pedsim_fp <- system.file("extdata", "GBR.sim.seg.gz", package="skater", mustWork=TRUE)
pedsim_seg <- read_ibd(pedsim_fp, source = "pedsim")
pedsim_seg
# $IBD1
# # A tibble: 1,553 × 6
# id1 id2 chr start end length
# <chr> <chr> <chr> <int> <int> <dbl>
# 1 testped1_g1-b1-s1 testped1_g2-b1-i1 1 752721 249170711 262.
# 2 testped1_g1-b1-s1 testped1_g2-b1-i1 2 118913 243043959 249.
# 3 testped1_g1-b1-s1 testped1_g2-b1-i1 3 108226 197800244 217.
# 4 testped1_g1-b1-s1 testped1_g2-b1-i1 4 167596 190936728 200.
# 5 testped1_g1-b1-s1 testped1_g2-b1-i1 5 157856 180692833 196.
# 6 testped1_g1-b1-s1 testped1_g2-b1-i1 6 183917 170981684 184.
# 7 testped1_g1-b1-s1 testped1_g2-b1-i1 7 46239 159119486 176.
# 8 testped1_g1-b1-s1 testped1_g2-b1-i1 8 113565 146280471 160.
# 9 testped1_g1-b1-s1 testped1_g2-b1-i1 9 212908 141027939 154.
# 10 testped1_g1-b1-s1 testped1_g2-b1-i1 10 158946 135473442 166.
# # ℹ 1,543 more rows
#
# $IBD2
# # A tibble: 132 × 6
# id1 id2 chr start end length
# <chr> <chr> <chr> <int> <int> <dbl>
# 1 testped1_g2-b1-i1 testped1_g2-b2-i1 1 156666011 162443758 9.43
# 2 testped1_g2-b1-i1 testped1_g2-b2-i1 1 197638290 213685761 20.5
# 3 testped1_g2-b1-i1 testped1_g2-b2-i1 1 243586697 249170711 9.43
# 4 testped1_g2-b1-i1 testped1_g2-b2-i1 2 40779973 67697179 25.7
# 5 testped1_g2-b1-i1 testped1_g2-b2-i1 3 26902677 27840868 0.797
# 6 testped1_g2-b1-i1 testped1_g2-b2-i1 3 186680562 192093520 12.1
# 7 testped1_g2-b1-i1 testped1_g2-b2-i1 4 81060970 100337853 16.7
# 8 testped1_g2-b1-i1 testped1_g2-b2-i1 5 24009109 30217553 4.83
# 9 testped1_g2-b1-i1 testped1_g2-b2-i1 5 31751157 134562539 83.7
# 10 testped1_g2-b1-i1 testped1_g2-b2-i1 5 167835827 168425497 1.15
# # ℹ 122 more rowsNotably, ped-sim differentiates IBD1 and IBD2 segments.
Given that IBD1 and IBD2 segments are weighted differently in kinship
calculation, this should be accounted for in processing. In the example
below the shared IBD is calculated separately for IBD1 and IBD2 with
type="IBD1" and type="IBD2" respectively. You
can then combine those results and sum the IBD1 and IBD2 kinship
coefficients to get the overall kinship coefficient:
ibd1_dat <- ibd2kin(.ibd_data=pedsim_seg$IBD1, .map=gmap, type="IBD1")
ibd2_dat <- ibd2kin(.ibd_data=pedsim_seg$IBD2, .map=gmap, type="IBD2")
dplyr::bind_rows(ibd1_dat,ibd2_dat) %>%
dplyr::group_by(id1,id2) %>%
dplyr::summarise(kinship = sum(kinship), .groups = "drop")
# # A tibble: 48 × 3
# id1 id2 kinship
# <chr> <chr> <dbl>
# 1 testped1_g1-b1-i1 testped1_g2-b1-i1 0.245
# 2 testped1_g1-b1-i1 testped1_g2-b2-i1 0.245
# 3 testped1_g1-b1-i1 testped1_g3-b1-i1 0.136
# 4 testped1_g1-b1-i1 testped1_g3-b2-i1 0.124
# 5 testped1_g1-b1-s1 testped1_g2-b1-i1 0.245
# 6 testped1_g1-b1-s1 testped1_g2-b2-i1 0.245
# 7 testped1_g1-b1-s1 testped1_g3-b1-i1 0.109
# 8 testped1_g1-b1-s1 testped1_g3-b2-i1 0.121
# 9 testped1_g2-b1-i1 testped1_g2-b2-i1 0.254
# 10 testped1_g2-b1-i1 testped1_g3-b1-i1 0.245
# # ℹ 38 more rows